
FDA生物制品的申报许可流程及要求
专栏:法规动态
发布日期:2018-01-05
阅读量:8311
2. The application of new biological products licensing process and requirements
Biological products as a special Drug, it gain FDA permission License (License) must be satisfied "with The Public Health Service Act" (The Public Health Service Act, PHS Act) part 351 (Section 351) on The biological mechanism and product licensing requirements, and The Federal Food, Drug and Cosmetic Act (The Federal Food, Drug, and Cosmetic FD&C Act) part 505 (Section 505) The requirements of The "new Drug".
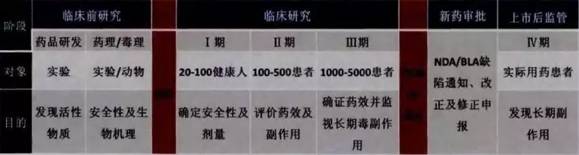
New Drug approval successful mainly takes preclinical study, research New Drug Application (IND, study New Drug), clinical research, New Drug applications (New Drug Application, NDA), the examination and approval and post-marketing surveillance phases (figure 2).
It is important to note that most of the biological medicine products are usually for Biologics license Application (Biologics license Application, BLA), only a few biological products (mainly restructuring hormones, such as insulin and human growth hormone, etc.) as a drug, for historical reasons for the NDA. Therefore, biomedical products BLA juxtaposed with NDA for two stage, declarer according to declare the properties of the different way of declaration. PHS Act regulations states in the United States sales of biological products must obtain a license.
When at the end of the third phase of clinical trial of new drug, the applicant may be proposed to the FDA for biologics license application (BLA) or new drug applications (NDA), in fully proved that the drug safety, efficacy and quality standards, can be issued by the drug certificate, the drug can enter new drug approval stage. Review of new drugs and biologics license application programs include: application of acceptance, new technology review, field investigation, inform the review result, bilateral exchanges (mid application submitted before the meeting, conference, end the review meeting and other meeting), etc.
New drug application is approved, in order to continue to ensure the safety and efficacy of drug, listed on the FDA to approve drug post-marketing surveillance. Therelease of post-marketing surveillance including biological products and post-marketing adverse event notification, biological products recall, etc. Manufacturers are required to more additional Clinical Trials, this is known as a new drug Clinical trial stage IV (Postmarketing Clinical Trials). While drug makers must also be timely to review and report to the FDA its knowledge every drug adverse events. Very serious and deadly drug adverse events must be reported within 15 days, other events are reported quarterly.
Figure 2 listed new drug research and development process
1553571941043073688 JPG
2.1. Research new drug (IND)
The FDA regulation begins with "of new drugs

2. The application of new biological products licensing process and requirements
Biological products as a special Drug, it gain FDA permission License (License) must be satisfied "with The Public Health Service Act" (The Public Health Service Act, PHS Act) part 351 (Section 351) on The biological mechanism and product licensing requirements, and The Federal Food, Drug and Cosmetic Act (The Federal Food, Drug, and Cosmetic FD&C Act) part 505 (Section 505) The requirements of The "new Drug".
New Drug approval successful mainly takes preclinical study, research New Drug Application (IND, study New Drug), clinical research, New Drug applications (New Drug Application, NDA), the examination and approval and post-marketing surveillance phases (figure 2).
It is important to note that most of the biological medicine products are usually for Biologics license Application (Biologics license Application, BLA), only a few biological products (mainly restructuring hormones, such as insulin and human growth hormone, etc.) as a drug, for historical reasons for the NDA. Therefore, biomedical products BLA juxtaposed with NDA for two stage, declarer according to declare the properties of the different way of declaration. PHS Act regulations states in the United States sales of biological products must obtain a license.
When at the end of the third phase of clinical trial of new drug, the applicant may be proposed to the FDA for biologics license application (BLA) or new drug applications (NDA), in fully proved that the drug safety, efficacy and quality standards, can be issued by the drug certificate, the drug can enter new drug approval stage. Review of new drugs and biologics license application programs include: application of acceptance, new technology review, field investigation, inform the review result, bilateral exchanges (mid application submitted before the meeting, conference, end the review meeting and other meeting), etc.
New drug application is approved, in order to continue to ensure the safety and efficacy of drug, listed on the FDA to approve drug post-marketing surveillance. Therelease of post-marketing surveillance including biological products and post-marketing adverse event notification, biological products recall, etc. Manufacturers are required to more additional Clinical Trials, this is known as a new drug Clinical trial stage IV (Postmarketing Clinical Trials). While drug makers must also be timely to review and report to the FDA its knowledge every drug adverse events. Very serious and deadly drug adverse events must be reported within 15 days, other events are reported quarterly.
Figure 2 listed new drug research and development process
2.1. Research new drug (IND)
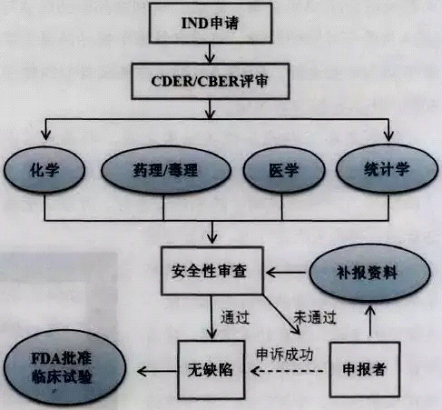
The FDA began to declare for new drugs supervision to submit application for IND, FDA will mainly IND applicants provide a clinical trial in vitro safety audit data and animal experiment, to determine whether the drug is safe enough to enter human trials. FDA is different according to the requirements of new drugs, the IND is divided into public permission of commercial IND and research (non-commercial) IND. Non-commercial IND application, and can be divided into researchers IND, based on the purpose of an emergency use IND and IND. The researchers IND generally put forward by the study doctor and dominate; Emergency IND general authorized by the FDA in case of an emergency use; Treatment IND in view of the serious threat of disease and has the quality outlook of experimental drugs, in the ongoing clinical trials at the FDA for approval at the same time.
Whether or not have clinical studies in other countries outside the United States, as long as it is in the United States to apply for the IND study, must fill in the FDA in 1571 and FDA form 1572. Application must provide declaration person name, contact information; Still need this research program, including research principles, indications, evaluation methods, is expected to clinical trial scheme, etc.; Declarant also need to provide a research manual, the manual used to assist in physicians to understand the research process. In addition, the application shall include the other three areas of information:
I. clinical research plan and information on the researchers. Detailed information such as clinical application as well as for the certification of the researchers.
Ii. The production information of the drug. Including components, impurity and stability, the source of drugs and the information such as quality control, etc.
Iii. The animal pharmacology and toxicology research. Preclinical animal testing data, you must abide by the good laboratory practice (GLP), not should abide by.
IND to apply for the required materials, and federal laws and regulations corresponding requirements as shown in table 1.
Table 1 IND of required materials and corresponding laws and regulations

Submit IND information, according to the regulation of classification the application materials will be assigned to the drug approval and research center (CDER) or biological product review and research center (CBER). CDER or CBER will IND review team, the review team including project managers, product/CMC (chemical production quality control) reviewers, pharmacology/toxicology reviewers, statistical reviewers, etc. Review team of key for reasonable security considerations. If the submitted materials without defect, the FDA must issue a reply within one month after receiving the application, otherwise said it had approved to enter human trials, automatically entered into clinical trials. IND application process is shown in figure 3.
图3 IND申请和审批流程
2.2 Clinical trials (Clinical trials)
Clinical trials for the purpose of the assessment research whether new drugs can be safely and effectively used to prevent or diagnosis of a particular disease or condition, the results will be one of the most important indicator of a drug approval. In new drug IND put forward by the applicant after review by the FDA approval, the proposed clinical trial plan (including the number of people participating in clinical trials with drugs and dosage, test process, test, etc.) and other relevant details needed to the ethics committee (Institutional review board, IRB) audit supervision, and ensure that participants are voluntary and must be told that their possible danger, and the researchers need to take the appropriate measures to protect people from harm.
Clinical trials is divided into four periods, each period to pass before he can enter the next phase, it is only through the first three periods are allowed to put forward new drug applications (NDA) or biologics license application (BLA).
I. Ⅰ period clinical (Phase I). In view of the healthy volunteers, main purpose is to observe the safety of new drugs on the human body, whether the side effects, drug dynamics in the early (Pharmacokinetics) and drug distribution, absorption, metabolism and excretion, etc.
Phase ii. Ⅱ (Phase ii). For a small number of patients, mainly to understand the curative effect of new drug, pharmacological study and determine the purpose, short-term toxicity and drug interaction, etc., to determine the effectiveness of the drug, at the same time, security and the side effects are also closely observe objects.
Phase iii. Ⅲ (Phase iii). For most patients, the purpose lies in the comprehensive study drug safety and efficacy. According to different human groups, different measurement, interactions with other drugs, and the side effects of long-term medication, etc. To carry out the test. This phase is the main stage of the new drug test, it is usually done in the second stage after should negotiate with the FDA (End of phase3 meeting), seeking FDA recommendations.
Phase iv. Ⅳ (Phase iv). After the approval of new drugs, is primarily to test the drug's long-term security, and whether for new species have side effects, etc.
2.3 new biological products and application
Upon completion of clinical trials, drug applicants shall not clinical research data, the results of the clinical trials, and the requirements of compliance documents submitted to the FDA audit, to determine whether approve the listing of new drugs. The FDA review all of the animals with human trials data, drug metabolism and dynamic data and drug GMP production data, etc.
A new biological products for licensing applications, on the basis of dividing property respectively can adopt different biologics license application (BLA) and new drug applications (NDA). Applicants to submit BLA or after NDA is accepted, then entered the stage of review. Biomedical products according to different attributes, submit CBER and CDER respectively
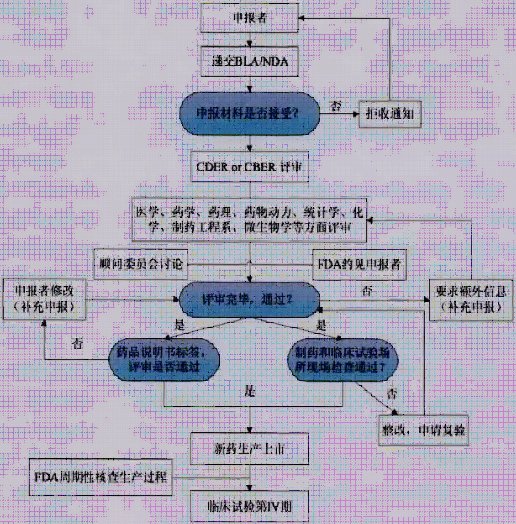
1) BLA
Most biomedical products take the BLA route. After completing the clinical trial, a BLA application can be made. In the case of a BLA application, the applicant is required to submit to the US FDA documentary materials to support the review and final approval of the drug's listing and sale in the United States, including raw materials, manufacturing processes, animal experiments, and clinical trials.
Applicants are required to submit an FDA 356h form and are required to submit the following information:
i. Applicant information. Including the applicant's name, address and phone number, the name and address of the producer;
Ii. Product/manufacturing information. Including raw materials, manufacturing and control processes, pollution/interaction information, environmental assessment, etc.;
Iii. All IND information;
Iv. all data from clinical trials (stages I-III);
v. side effects;
Vi. Product labels, etc.
For the information provided, the FDA's approval procedures will primarily include reviewing all information in the IND application, including pre-clinical studies (animal tests), clinical studies (human trials) data; evaluating clinical trial results and analysis; reviewing label content And FDA on-site inspections through production facilities and major clinical trial sites.
After the approval of the BLA, the following requirements must also be met:
i. Annual Report: PMC Status (21 CFR 601.70);
Ii. Other materials to be submitted: side effects (21 CFR 600.80), sales summary (21 CFR 600.81), biological abnormal report (21 CFR 600.14), and advertising and promotional labels;
Iii. Periodic cGMP audit.
In addition, in accordance with 21 CFR 601.12, after the BLA has passed the audit, it should be reported to the FDA in a timely manner whenever production and label changes are involved.
2) NDA
At present, there are still a small number of biological products, such as insulin, glucagon, human growth hormone, etc., declared by NDA. After completing the third phase of the clinical trial, the applicant must provide all the animal, human test data and data analysis results in addition to the effects of the drug on the human body and the manufacturing process of the drug. When an NDA application is submitted to the FDA, the FDA has 60 days to decide whether to file the application for further review, or to reject applications that are incomplete or missing certain test items. The approval standard of NDA is whether the new drug is safe and effective according to the expected use, whether the benefit of the drug is far greater than the risk; whether the label of the new drug is suitable, what should be; what is the quality and quality of the drug manufacturing and quality control process? And purity.
NDA can be written in accordance with the NDA requirements of 21 CFR 314.50, or the General Technical Document (CTD) model established and widely implemented by ICH. The Drug Archive (DMF) is also part of the NDA's partial data declaration to support IND, NDA and ANDA. The FDA is currently encouraging the use of electronic filing.
The forms and concerns that NDA needs to submit include the FDA 356h form (applying for commercial new drugs, biopharmaceuticals or antibiotics), the FDA 3397 form (fees), and the FDA 3331 form (new drug application area report). Its main content covers administrative management and prescription information, CTD declaration content and format outline, quality, non-clinical research reports and clinical research reports.
Similar to the FDA's main approval procedures for BLA as described in the previous section, the FDA's approval of NDA submissions also includes the following aspects:
i. All information in the Investigative New Drug (IND) application, including all preclinical studies (animal tests), clinical studies (human trials) data;
Ii. Results and analysis of clinical trials;
Iii. the content of the label;
Iv. FDA on-site inspections through production facilities and major clinical trial sites.
3. Type of review
In 1992, under the Prescription Drug User Act (PDUFA), the FDA agreed to speed up drug reviews and developed two review cycle systems, Standard Review and Priority Review.
The standard review applies to most drugs that have a slight improvement and improvement over the efficacy of commercially available drugs. The 2002 revision of the PDUFA requires that the standard review of new drug applications be completed within a 10-month timeframe. Priority review is applicable to drugs that are significantly more therapeutically remedy than those already on the market, or that provide a treatment that does not exist on the market. The priority review greatly shortens the review time, and the target period for completing the review is 6 months.
Because the FDA is aware that the time to obtain clinical data can be long, sometimes it can take up to several years. As a result, the FDA established Accelerated Approval in 1992 to allow early adoption of drugs used to treat major diseases. Drug applicants need to submit a message based on the Surrogate endpoint analysis method. An alternative endpoint is an indicator or surrogate measure of an experiment in a clinical trial that is used to represent a meaningful clinical outcome such as survival or improvement in symptoms. The use of alternative endpoints can significantly reduce valuable time in the process of drug approval. For example, the FDA can wait until the drug does extend the data on the survival time of the tumor patient before making a judgment. Instead, the drug can be reviewed based on evidence that the tumor has become smaller after the drug treatment, because this evidence can Reasonably speculate on the actual clinical effect. After the FDA passed the drug review, drug manufacturers still need to continue to verify whether the evidence of tumor shrinkage in clinical IV is associated with prolonged survival of cancer patients. If the fourth phase of the data confirms the inference, the FDA will grant the drug approval in the traditional mode; if the Phase IV results are insufficient to support the inference, the FDA will initiate a regulatory process that may order the drug to be delisted. The accelerated approval model saves time from drug development to time-to-market and is beneficial for drug interviews that potentially treat major illnesses.
In addition, FDA approval also has a "Fast Track" approach to address products and requirements that have not met unmet medical needs, that is, those that are not yet available or potential in the market are better than those already on the market. Development and approval of new drugs with drugs that have the potential to treat serious and life-threatening diseases. Applicants must have better efficacy than existing drugs, avoid the serious side effects of existing therapies, improve the diagnosis of key diseases, and significantly reduce clinical toxicity. Meeting these requirements can be assigned as a rapid pathway. In addition, most drugs that are assigned as a fast route are likely to be recognized by the FDA for priority review. Drug companies can apply for a quick route to drugs at any time during the development process. The FDA will respond to the company's application within 60 days.
4. Approval of Biosimilars
Biosimilars refer to biological products that are highly similar to approved biological products (reference products/original products). Despite the subtle differences in clinically inactive ingredients, there is no clinically significant difference in safety, purity, and efficacy between biosimilars and approved biologics.
In 2010, the Biological Product Price Competition and Innovation Act (BPCI Act) came into force as part of the Health Care Reform Act of the Affordable Care Act, which amended the PHS Act and developed 351. Section (k) granted the FDA the right to approve biosimilars and established a biosimilars to simplify the application process. Biosimilars can be applied as long as they demonstrate biosimilarity or interchangeability with FDA-approved biologics.
The licensing process for biosimilars is similar to most biopharmaceutical products. The difference is that the submission of biosimilars focuses on demonstrating universal or biological similarity to biomedical products (reference products) that have been approved by the FDA.
The application information submitted by 351(k) includes at least:
i. Prove that it is biologically similar to the original product;
Ii. Under the assumption of the same conditions of use, it proves that it has the same mechanism of action as the original product;
Iii. The conditions of use indicated on the generic label have been approved in the original product;
Iv. Prove the same route of administration, dosage and strength as the original product.
Evidence to demonstrate biosimilarity comes from analytical studies, animal studies, and clinical studies. Among the analytical studies, although there are subtle differences in clinical inactive ingredients, the analytical data can demonstrate a high degree of similarity between biosimilars and approved biologics; animal studies must include toxicological assessment; in clinical studies, Under one or more conditions of use of the original drug, the evaluation of the immunogenicity, PK, and PD of the generic drug is sufficient to prove that the safety, purity, and efficacy are compared, but complete clinical trial data is not required.
According to the Health Care Reform Act, biosimilars applicants are not allowed to submit a simplified application for biosimilars to the FDA within four years of the new drug being approved for sale, and the FDA cannot approve a simplified application for biosimilars within 12 years of the new drug being approved for sale. This ensures that the market for new drugs will be exclusively for at least 12 years. At the same time, the simplified application route establishes a mechanism for patent exchange between the manufacturer of the new drug and the biosimilar, that is, the applicant of the biosimilar is required to provide the new drug manufacturer with a new drug manufacturer within 20 days of receiving the FDA acceptance application. Make a copy of the application and disclose its production process. Within 60 days of receiving the information, the new drug manufacturer must provide the generic drug applicant with a list of patents listing all patents for generic infringement and indicating to the other party that they are willing to license the patent. Applicants for generic drugs must provide a statement within 60 days of receipt of the patent, detailing why the patent list listed is invalid, not enforceable or not infringed by the generic product, otherwise the generic applicant must declare There is no commercial marketing of generic drugs until the patent expires. In response to this statement, the manufacturer of the new drug must specify within 60 days why the listed patent is valid, enforceable and infringed by generic drugs.
5. Summary
The United States is the fastest growing biopharmaceutical industry in the world, and has a strict and complete operational mechanism for the entire process of reporting and reviewing new biologics and biosimilars. The market access requirements for biomedical products developed by the FDA and the approval indicators for biological products largely reflect the advancement of technology and the highest standards of quality, safety and efficacy.